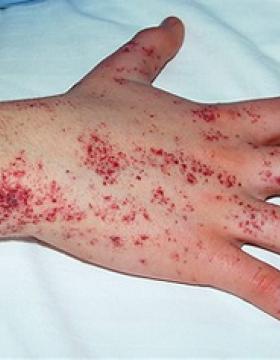
La Hemofilia A o déficit de Factor VIII es la forma más frecuente de hemofilia, afecta a 1 de cada 6000 individuos del sexo masculino. Es una patología que afecta a la coagulación sanguínea que se caracteriza por hemorragias espontáneas o prolongadas.
Se debe a una alteración del cromosoma X, es una patología recesiva; por lo tanto, las mujeres actúan casi siempre de portadoras y los hombres son los que padecen la enfermedad. Los casos de Hemofilia en mujeres son muy poco frecuentes pero sí que hay algunos casos documentados, como el de la Reina Victoria I.
La enfermedad suele aparecer cuando los niños afectados empiezan a andar. La gravedad de las manifestaciones depende de el déficit de Factor VIII de coagulación.
El tratamiento, básicamente, es sustitutivo. Es frecuente que los anticuerpos ataquen al factor de coagulación administrado, inhibiendo su acción. Es una enfermedad grave que, históricamente, ha causado muchas muertes aunque los nuevos tratamientos le dan un enfoque más favorable.
Hay varios tipos de Hemofilia A:
Actividad biológica del Factor VIII menor al 1%: hemorragias de forma espontánea y frecuentes. Sangrados anormales como resultado de heridas leves.
Actividad biológica del Factor VIII entre 1% y 5%: sangrados anormales como resultado de heridas leves. Las hemorragias de forma espontánea no son frecuentes.
Actividad biológica del Factor VIII entre 5% y 40%: sangrados anormales como resultado de heridas leves.
A continuación, os dejo un enlace para que tengáis más información:
Hoy os hablaré del síndrome de apert que está caracterizado por el cierre prematuro de las suturas
craneales que a su vez, ocasiona malformaciones en el cráneo, cara, manos y pies
así como alteraciones funcionales que difieren de unos niños a
otros. Se da en 1 de cada 100.000 nacimientos.
¿Cuál es su causa?
Se debe a una
mutación en el genFGFR2que provoca que
algunas de las suturas óseas del cráneo se cierren antes de tiempo
(cráneosinotosis).
Puede transmitirse
de padres a hijos como rasgo autosómico dominante aunque en algunas
casos al tratarse de una mutación se produce sin haber ningún
antecedente conocido.
¿Cuál es su sintomatología?
Los síntomas más
comunes de esta enfermedad son:
Deformación
esqueléticas como fusión de los dedos de las manos y los pies
(oséa o fel tejido subcutáneo)
Cráneosinotosis
que si no se trata puede provocar :
Hipertensión
craneal
Retraso
mental
Ceguera
Pérdida de
audición
Apariencia
característica de la cara con subdesarrollo grave de la parte media
de la cara
Ojos
prominentes
Dificultades
en el cierre de la arcada superior e inferior
Baja estatura
Pérdida de
la audición de los oídos
¿Cuáles son sus expectativas y tratamiento?
El pronóstico de
esta enfermedad es muy variable en cada niño, pero se recomienda a
los padres potenciales acudir a asesoría genética y realizar las
cirugías lo antes posible. Como ya adelanté,
el tratamiento se basa en cirugías para “reparar” el crecimiento
anómalo de los huesos del cráneo y de las extremidades fusionadas. En casos de problemas auditivos se debe acudir a un
otorrino.
En una anterior entrada he hablado sobre un tipo de leucemia, en concreto, sobre la leucemia mieloide crónica. Esta entrada, se la voy a dedicar a otro tipo de leucemia: la leucemia linfocítica crónica (LLC).
La LLC es también una enfermedad hematológica rara que se suele presentar en mayores de 50 años afectando más a los varones y que se produce cuando hay una acumulación de linfocitos de aspecto normal en sangre periférica y que se infiltran en médula ósea, bazo y ganglios linfáticos.
Los síntomas de este tipo de leucemia son poco llamativos, estando clínicamente caracterizado por adenopatías (inflamaciones de los ganglios), infecciones de repetición, esplenomegalia y/o hepatomegalia y síntomas característicos de anemia como la astenia (debilitación del estado general), palidez, palpitaciones y mareos.
De este modo, los pacientes con esta enfermedad avanzada pueden llegar a presentar anemia, granulocitopenia y trombocitopenia, por estar infiltrada la médula ósea con células neoplásicas. Además, el 20% de estos enfermos, desarrolla una anemia hemolítica autoinmune.
En cuanto al pronóstico de la leucemia linfocítica crónica, éste está relacionado con la etapa de la enfermedad en la que se encuentre el paciente:
Etapa A. Se caracteriza por linfocitosis con afectación de menos de tres grupos de ganglios linfáticos. En esta etapa, el paciente tiene un buen pronóstico de supervivencia, siendo la media superior a siete años.
Etapa B. Con afectación de más de tres grupos ganglionares y hepatoesplenomegalia. El paciente en esta etapa, tiene un pronóstico intermedio con una supervivencia aproximada de cinco años.
Etapa C. Cualquier estadío evolutivo que presente anemia o trombocitenia. El paciente tiene un mal pronóstico con una etapa de supervivencia inferior a dos años.
El tratamiento de la LLC, como el de las demás leucemias en general, puede ser de varios tipos: quimioterapia, uso de medicamentos que eliminan las células cancerosas; radioterapia, uso de rayos X u otros de alta energía que eliminan a estas células y reducen tumores; inmunoterapia o terapia biológica, tratamiento que intenta que el propio organismo combata el cáncer; y trasplante de médula ósea, reemplazamiento de la médula ósea afectada por una sana.
A continuación, os dejo un vídeo de un informativo en el que se explica el nuevo descubrimiento que ha hecho un equipo de investigación y cómo esto puede ayudar a los pacientes creando un tratamiento personalizado:
Para más información: http://www.enfermedades-raras.org/
Como vimos en la anterior entrada, la hidrocefalia es un acúmulo anormal de LCR en el cráneo.
Hoy hablaremos un poco de cuales son las pruebas diagnósticas y el tratamiento de esta afectación
¿Pruebas diagnósticas?
Mediciones repetitivas del perímetro cefálico pueden mostrar que la cabeza se está engrandando
Una tomografía computarizada de la cabeza es el mejor examen para detectarla.
Arteriografía
Gammagrafía cerebral
Ecografía y radiografía del cráneo
Punción lumbar y análisis del LCR
¿Y cuál es su tratamiento?
Nuestro objetivo principal es reducir y prevenir el daño cerebral mejorando el flujo del LCR.
Siempre que se pueda, se puede realizar una cirugía para eliminar la obstrucción.
En el caso de que no se pueda realizar la cirugía, podemos colocar una sonda flexible que se llama "DERIVACIÓN" dentro del cerebro para redireccionar el flujo del LCR a otra parte del cuerpo como el abdomen para su posterior absorción.
Existen otros tratamientos:
Antibióticos: si hay infección.
Ventriculostomía endoscópica del tercer ventrículo (ETV), que libera presión
Extrirpación de las partes del cerebro que producen LCR
Se necesitan chequeos regulares regulares para constatar que no existan problemas posteriores.
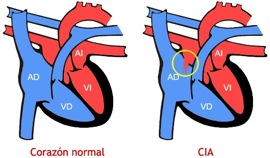
Hoy os voy a hablar sobre la comunicación interauricular (CIA) que es otro de los tipos de cardiopatía congénita. Aunque en otra entrada ya he hablado sobre la comunicación interventricular, estas dos patologías afectan de distinta forma al sistema cardiovascular.
En la CIA, existe una abertura que comunica la parte derecha e izquierda del corazón mediante las aurículas (su parte superior) y, normalmente, no se hereda genéticamente siendo la causa usualmente desconocida.
En cuanto al trabajo que tiene que realizar el corazón, sí que es igual a la comunicación interventricular y, por lo tanto, si la abertura es grande el corazón debe de trabajar más de lo habitual siendo la presión y el flujo de la sangre enviada a los pulmones mayores de lo normal. Esto puede derivar en una congestión pulmonar y, esto a su vez, puede dar lugar a un deterioro del intercambio gaseoso y/o a una hipoxemia arterial.
La CIA puede curarse normalmente mediante una intervención quirúrgica en la infancia. De este modo, se evitan problemas de corazón y pulmonares en la edad adulta. Sin embargo, cuando la comunicación es pequeña, se puede cerrar de forma espontánea o mediante la colocación de un dispositivo a través de un cateterismo cardíaco, sin necesidad de abrir el tórax.
¿Cómo afecta a los niños?
Si la CIA es grave, se cansarán fácilmente. Además, algunos niños padecerán resfriados e incluso bronconeumonías frecuentes. Por el contrario, otros niños no padecerán síntomas.
La enfermedad de Lowe o Distrofia Cerebro Oculorenal es una enfermedad rara que afecta a 1 de cada 500000 personas. Es un trastorno multisistémico que causa anomalías en los riñones, los ojos y el sistema nervioso. Está ligada al cromosoma X y afecta, principalmente, a los varones.
Los bebés que nacen con este síndrome nacen ya con catarata bilateral e hipotonía grave. En las semanas siguientes al nacimiento, se hace evidente la tubulopatía renal proximal; pueden aparecer queloides o glaucoma en los ojos, hipotonía, convulsiones...Padecen un retraso psicomotor severo.
No tiene cura pero los síntomas y signos asociados al síndrome se intentan paliar: extracción de cataratas, terapia física y del lenguaje...
Seguramente que
alguna vez hayáis visto a una persona con una “mancha” enorme por
el cuerpo o la cara, no es una mancha, si no que se trata de un lunar
gigante conocido como nevus.
No se trata de una
enfermedad rara en sí, pero si que que es una malformación bastante
atípica que me ha parecido bastante interesante y que creo que a
vosotros también os podría interesar.
Como
ya os dije, un nevus gigante congénito
es un lunar muy
grande presente en el momento del nacimiento. La comunidad médica aun
no se ha puesto de acuerdo sobre lo grande que tiene que ser un lunar
para considerarlo
como
"gigante", y además,
los
criterios varían según la zona del cuerpo en la que esté presente
y la edad del afectado.
Debemos
tener en cuenta que un nevus gigante es congénito, pero no
hereditario, es decir, la malformación se produce durante el
desarrollo embrionario,en algún momento de la vida uterina del niño
se produce un "error de fabricación" que interfiere con el
desarrollo y dispersión de los melanocitos. Como no es hereditario,
no se transmite de padres a hijos, por lo que es muy raro que se
repita entre hermanos.
La
causa del nevus gigante congénito es desconocida y no se conoce
ningún factor que pueda ser responsable de esta malformación.
Los nevus
no se "curan" pero
se
pueden extirpar total o parcialmente, o si
no, se pueden dejar
estar, pero en cualquiera de los casos es importante un control
médico regular y una visita regular al dermatólogo, pues
como lunar que es, debe tratarse con cuidado y con ciertas
precauciones.
Es una condición en la que los ventrículos cerebrales,
también llamados espacios cerebrales, está dilatados anormalmente e impiden el
flujo normal del líquido cerebroespinal
La hidrocefalia significa del agua en el cerebro.
¿Cómo se produce este caso?
El líquido cerebroespinal o liquido cefalorraquídeo se
acumula en el cráneo y somete a un aumento de presión a los tejidos del
cerebro.
El liquido cefalorraquídeo (LCR) normalmente circula a
través del cerebro y la médula espinal y se absorbe en el torrente sanguíneo.
Demasiado LRC ejerce presión sobre el cerebro, lo cual lo empuja hacia arriba
contra el cráneo y daña el tejido cerebral.
¿A que edad se puede manifestar?
Ya se puede encontrar mientras el feto está creciendo dentro
del útero materno. Es más común en bebés que presentan mielomeningocele
(defecto en la columna vertebral que no se cierra de manera correcta).
¿Cuál es su sintomatología?
Dependen de la edad, la cantidad del daño cerebral y lo que
está causando dicho acumulo de LCR. Destacamos:
Vómitos
Somnolencia
Irritabilidad
Y en los bebés:
Llanto breve, chillón y agudo
Dolor de cabeza
Dificultad para comer
Pérdida de la coordinación
Este se puede manifestar con la presencia de venas
hinchadas, sonidos anormales cuando el médico golpea el cráneo del niño o la
presencia de ojos hundidos.
Hoy, os voy a hablar de la enfermedad del hombre lobo, la hipertricosis. Es una patología que se caracteriza por un crecimiento excesivo de pelo en cualquier parte del cuerpo. Puede ser localizada, si el crecimiento anómalo de pelo ocurre en una parte concreta del cuerpo, o generalizada, si ocurre en todo el cuerpo.
Se debe a una mutación genética del cromosoma X.
Esta patología no es una enfermedad en sí misma ya que el único problema que causa es estético.
El tipo de hipertricosis más grave es la hipertricosis lanuginosa congénita: aquellas personas que la padecen tienen todo el cuerpo cubierto de pelo excepto las plantas de los pies y las palmas de las manos. Es extremadamente rara ya que desde la Edad Media solo se han documentado unos 50 casos.
Si alguna vez
pensáis que estáis teniendo un ataque al corazón ¡que no cunda el
pánico! podría existir la posibilidad de que se estuviera tratando
del síndrome de tietze! Aún así no estaría de más ir a urgencias lo antes posible por si las moscas..
Como ya sabréis
hoy os hablaré del síndrome te tietze, una enfermedad rara
caracterizada por la inflamación del cartílago que se encuentra
entre las costillas y el esternón, el cartílago costal, al
inflamarse produce bastante dolor lo que podría confundirse con el de
un ataque al corazón. Este síndrome afecta a hombres y mujeres por
igual y es bastante más común en personas jóvenes de entre 20 y 40
años que en personas mayores de los 40 años.
¿Cuál es su causa?
La causa de este
síndrome es desconocida, pero se suele relacionar con el esfuerzo
físico o lesiones producidas por estornudos y toses persistente,
infección de las vías respiratorias o incluso por un buen ataque de
risa, por otra parte, se piensa que el estrés o los tratamientos de
radioterapia prodían ser agravantes de esta enfermedad.
¿Cuál es su
sintomatología?
Sus síntomas más
comunes son:
Dolor de
carácter mecánico e intensidad variable, pudiendose confundir como
ya hemos dicho con dolor en el corazón. El dolor puede durar desde
horas a semanas y
Inflamación
en la zonas de las costillas y pecho.
Enrojecimiento
y sensibilidad al calor
¿Cuáles son sus
expectativas?
Tiende
a resolverse de forma espontánea después
de
dos meses pero puede
prolongarse
y de forma muyinusual
se mantiene de forma crónica. No es
frecuente
requerir tratamiento pero
se aconseja
evitar cualquier movimiento que empeore los síntomas.
¿Como
se trata el síndrome de tietze?
El síndrome de
tietze se puede tratar con antiinflamatorios (no esteroideos),
fisioterapia respiratoria, inyecciones de cortisona (para tratar el
dolor), parches de licodaína o la aplicación de bolsas de hielo en
la zona inflamada.
Se trata de una malformación congénita que tiene las mismas características tanto en hombres como en mujeres.El recto está completamente ciego y suele encontrarse unos 2 cm por encima de la piel perineal.
¿Qué causas presenta?
Puede ocurrir:
El recto puede terminar en una bolsa ciega que no se conecta con el colon.
Puede haber estrechamiento o ausencia del ano.
Es causado durante la etapa fetal.
¿Y su sintomatología?
Orificio anal muy cerca de la vagina.
El bebé no es capaz de eliminar la primera deposición.
Presencia de una área abdominal hinchada.
Ausencia del orificio anal.
Se asocia frecuentemente con el Síndrome de Down.
¿Tiene tratamiento?
Se requiere cirugía para la corrección del ano y en el caso de que el recto se encuentra en contacto con otros órganos, se tendrá que realizar la reparación de los órganos dañados.
Lo más frecuente es que se realice una colostomía temporal que consiste en conectar el extremo del intestino grueso en la pared abdominal de manera que las heces sean recolectadas en una bolsa que se coloca en dicha zona.
Hoy, voy a hablaros sobre la enfermedad de Wilson, también conocida como degeneración hepatolenticular o degeneración lenticular progresiva.
Esta enfermedad es un trastorno autosómico recesivo, que se caracteriza por la acumulación tóxica de cobre en el hígado y en el sistema nervioso central, principalmente. El diagnóstico va a depender de la detección de los signos clínicos y fenotípicos propios de la enfermedad y, además, de la detección de anomalías genéticas asociadas.
Los pacientes pueden presentar cuadros hepáticos, neurológicos o incluso psiquiátricos. Estos pacientes, pueden ser tratados de forma eficiente usando quelantes de zinc, pero el tratamiento es muy caro.
Por otro lado, hay que destacar que la terapia recomendada en casos de pacientes con hepatitis fulminante o con una progresión continua de la disfunción hepática sin respuesta a la terapia farmacológica, anteriormente citada, es el trasplante de hígado.
A continuación, os dejo un vídeo sobre un paciente que sufre esta patología:
Es una falta congénita del iris del ojo. En estos casos puede ser total o parcial, y normalmente se tiene en ambos ojos. Junto con esta enfermedad suelen surgir complicaciones como: cataratas, nistagmus, glaucoma. Existen otras más importantes como afecciones renales o retraso mental.
¿Cuál es su sintomatología?
Presentan una gran fotofobia debido a la falta del iris
Agudeza visual inferior a un 20%
¿Puede ser hereditario?
Sí, es una enfermedad congénita y hereditaria. Se transmite entorno a un 50% de los descendientes de una forma autosómica dominante. Es producida por una deleción en el gen PAX 6, responsable de la formación del globo ocular durante la gestación, en el cromosoma 11.
¿Y su tratamiento?
Es muy complicado, ya que las gafas convencionales no suelen mejorar la visión del paciente por lo que necesitan ayuda de baja visión. Las lentillas cosméticas con iris pigmentado alivian la fotofobia pero no mejora la agudeza visual.
Las intervenciones quirúrgicas se están empezando a utilizar para la introducción de implantes de lentes intraoculares con un iris artificial.
Esta entrada se la voy a dedicar a la enfermedad de Fabry debido a que mañana dará comienzo el VI Congreso Nacional dedicado a la enfermedad de Fabry organizado por la asociación MPS-Fabry España en Barcelona.
La enfermedad de Fabry, también conocida como déficit de ceramida trihexosidasa o lipodosis hereditaria distópica, es una enfermedad genética muy rara. Además, esta patología se incluye dentro del grupo de enfermedades metabólicas hereditarias de depósito lisosomal. Una vez dicho esto, hay que aclarar que un lisosoma es una estructura de la célula que funciona como las unidades digestivas elementales.
La causa de la enfermedad de Fabry es el déficit de un enzima lisosomal (alfa galactosidasa), presente en muchos tipos de células del organismo, y que está implicada en la rotura de la globotriaosilceramida (Gb3). Por lo tanto, esta enzima va a estar parcial o totalmente inactiva en esta patología.
Como consecuencia, la Gb3 se va a acumular en las células del organismo y va a alterar las funciones de diferentes órganos diana, como los riñones, el corazón, el sistema nervioso y la piel, siendo estos los más afectados. Los síntomas principales serán: lesiones cutáneas muy típicas (angioqueratomas), disminución o ausencia de sudoración, opacidades en córnea y cristalino y dolor y parestesias en extremidades con afectación vascular de cerebro, corazón y riñón.
Hoy hablaremos del pénfigo, una enfermedad rara autoimune en la que se producen
anticuerpos contra la propia piel y mucosas lo que origina :
ampollas de gran tamaño, separación de las células de la piel o
acumulación de líquido entre las distintas capas de la dermis.
Causa
Actualmente
no se conoce el motivo de esta enfermedad, pero ,según los estudios, no parece ser hereditario, únicamente hay personas con genes que los
hacen más vulnerables a padecer dicha enfermedad. Se sabe también que no es una enfermedad contagiosa.
Síntomas y tipos
Aproximadamente el 50% de las personas primero desarrollan ampollas dolorosas y
úlceras que drenan, supuran, forman costra o se desprenden fácilmente,
en la boca, cuero cabelludo o en el tronco, seguidas de ampollas en la
piel. Las úlceras cutáneas pueden aparecer y desaparecer.
Expectativas
y tratamiento
Por
lo general, el pénfigo responde bien al tratamiento, después de
varios años los pacientes pueden dejarlo aunque a veces puede
reaparecer.
El
tratamiento consiste en la utilización de altas dosis de
medicamentos antiinflamatorios (corticoesteroides) para controlar el
sistema inmunitario que también pueden ser usado en las ampollas.
Estos medicamentos pueden tomarse por vía oral, tópica, o pueden
inyectarse.
Otros
medicamentos que se pueden emplear son: los inmunodepresores para
controlar el sistema inmunitario y los antibióticos para tratar
posibles infecciones.
También recibe el nombre de Trisomía 18 ya que es una enfermedad cromosómica rara que se caracteriza por tener un cromosoma adicional en dicho par. Se presenta en 1 de cada 6000-13000 nacimientos y es 3 veces más común en niñas que en niños. Aumenta la incidencia de casos de la mano de la edad materna.
Se puede descubrir la patología durante el embarazo ya que las madres que gestan bebés con este síndrome tienen un útero inusualmente grande y mucho líquido amniótico.
Los afectados presentan talla corta, retraso mental y del desarrollo psicomotor e hipertonía. Además, presentan anomalías cardíacas, cráneo-faciales, oculares, esqueléticas, urogenitales, gastrointestinales, del sistema nervioso...
Sufren con mucha frecuencia neumonías, otitis, infecciones urinarias y problemas para comer (necesitando sonda en muchos casos).
Es una enfermedad con un pronóstico realmente malo ya que el 95% de los afectados fallece en el primer año de vida (debido, casi siempre, a una neumonía o a las malformaciones cardíacas).
Como siempre, os dejo un par de enlaces por si queréis buscar más información sobre esta patología:
Es un término utilizado para denominar a un grupo de enfermedades metabólicas en las que se acumulan proteínas anormales en tejidos y órganos. La acumulación de las proteínas anormales se denominan depósitos amiloideos. Su acumulación puede ser localizado o generalizado o sistémico.
¿Cuáles son sus causas?
Su causa es desconocida. Está relacionada con la producción anormal y excesiva de anticuerpos por parte de las células inmunitarias. Las proteínas anormales se acumulan en ciertos órganos impidiendo que estos trabajen correctamente. Se compara con la mieloma múltiple.
¿Que síntomas presenta?
Sus síntomas dependen principalmente de los órganos afectados, aunque abarcan:
Fatiga
Dificultad respiratoria
Pérdida de peso
Ritmo cardíaco anormal
Hinchazón en la lengua
¿Y de su tratamiento?
Se trata igual que el mieloma múltiple el cual incluye quimioterapia y transplante de células madre.
Si esta es causada por otra enfermedad, se debe tratar de una forma muy agresiva.
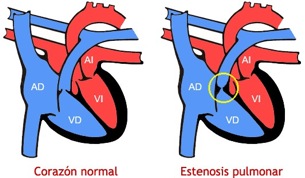
Hoy, os voy a dedicar esta entrada a hablar sobre la estenosis pulmonar (EP). Al igual que la coartación de la aorta, es uno de los tipos más "comunes" de cardiopatía congénita, y digo "comunes" porque sigue siendo una enfermedad rara.
La EP se debe a que se produce un ensanchamiento de la válvula tricúspide. La causa normalmente es desconocida y, generalmente, no se hereda genéticamente.
En este tipo de cardiopatía, el ventrículo derecho tendrá que trabajar más para poder impulsar la sangre a través de la válvula tricúspide estrechada. Además, puede ser curada mediante una intervención quirúrgica, si el estrechamiento es severo; o dependiendo del caso, a través de un catéter balón para poder abrir la válvula tricúspide sin necesidad de tener que sufrir una intervención invasiva.
¿Cómo afecta a los niños?
El niño se cansará fácilmente al realizar ejercicio y podrá presentar incluso cianosis, cuando el estrechamiento sea severo, porque llega poca sangre a los pulmones para que se oxigene.
Enfermedad
rarísima genética hereditaria que afecta al tejido
conectivo,esqueleto, pulmones, ojos, corazón y vasos sanguíneos.
Esta caracterizada por la presencia de malformaciones óseas, dedos
extra largos y delgados, pecho hacia fuera o hacia adentro,
hiperextensibilidad de las articulaciones, rodillas y piernas
curvadas hacia atrás, pies planos, y curvatura anormal de la
columna. Puede afectar a hombres, mujeres y niños. Se encuentra en
personas de todas las razas y grupos étnicos.
¿A
qué se debe?
El
síndrome de Marfan se debe a defectos presentes en un gen llamado
fibrilina-1
implicado en la producción de fibrilina, proteína necesaria para la
formación del tejido conectivo, que cuando es escasa o defectuosa
provoca el debilitamiento de este tejido. La anomalía en este gen
también causa que los huesos largos del cuerpo se desarrollen
demasiado, por lo que los pacientes presentan una estatura elevada
,piernas y manos largas.
El
tejido conectivo está presente en casi todo el organismo por lo que
la enfermedad afecta a muchísimos órganos y sistemas corporales
como el corazón, los pulmones, los ojos, los vasos sanguíneos
(especialmente la aorta), la piel, el tejido que cubre la médula
espinal y los huesos.
¿Cuáles
son sus síntomas?
Las
personas con el síndrome de Marfan son altas con brazos y piernas
delgados y largos, dedos en forma de araña (aracnodactilia) y
articulaciones demasiado flexibles.
Con
un tratamiento adecuado los enfermos tienen una esperanza de vida
normal, es importante un diagnostico precoz por que la mayoría de
los síntomas pueden ser tratados.
Para
disminuir la presión arterial se suelen prescribir betabloqueantes
que disminuyen la fuerza, frecuencia de los latidos y su incidencia
sobre la pared de la aorta evitando su ruptura.
La
mayoría de los problemas visuales como la miopía, suelen
solucionarse con la utilización de gafas. En caso de cataratas o
glaucoma un tratamiento temprano puede atenuar el deterioro de la
vista.
A
los pacientes de esta enfermedad se les avisa sobre los riesgos del
estrés físico o emocional intenso, ya que puede exigir un
sobreesfuerzo a la aorta.
Hasta aquí el síndrome de Marfan, si queréis más información:
La Adrenoleucodistrofia es una enfermedad metabólica hereditaria rara. Consiste en la acumulación, tanto en los tejidos como en los líquidos corporales, de ácidos grasos saturados de cadena larga. Los enfermos van sufriendo una degeneración progresiva de la corteza suprarrenal, lo que provoca una insuficiencia suprarrenal.
Se diferencian 3 tipos de adrenoleucodistrofia:
Neonatal: Comienza en los primeros meses de vida o en el periodo neonatal. Los lactantes tienen un deterioro neurológico y signos de disfunción de la corteza suprarrenal, retraso mental y suelen fallecer antes de los 5 años de vida. Se hereda como un rasgo genético autosómico recesivo.
Infantil: Comienza en la infancia o en la adolescencia. La degeneración neurológica avanza hasta una demencia grave y se va perdiendo la vista, el oído, el habla, la marcha..., hasta el fallecimiento. Se hereda como un rasgo genético ligado al cromosoma X (Enfermedad de Schilder).
Adulta: Comienza al final de la adolescencia o al principio de la edad adulta. Pueden pasar muchos años hasta que aparezcan los síntomas neurológicos. Ligada al cromosoma X pero más leve que la infantil.
La adrenoleucodistrofia infantil es la más frecuente, afectando a 1 de cada 35000 nacimientos.
Es una enfermedad que no tiene cura; salvo excepciones que, con un transplante de médula, se curan o mejoran. El tratamiento dietético es fundamental, se deben restringir las grasas saturadas.
También se usa un aceite denominado aceite de Lorenzo (muy útil si se utiliza antes de que aparezcan los síntomas neurológicos). Recibe este nombre porque fue descubierto por Augusto y Michaela Odone, padres de Lorenzo, un niño que fue diagnosticado de adrenoleucodistrofia en 1984. Los padres de Lorenzo lucharon mucho por intentar encontrar una cura para su hijo (siendo economista y lingüísta, nada que ver con la sanidad, la biología o la investigación) y, finalmente, consiguieron lo más parecido a una cura. Es un tratamiento caro que, hoy en día, muchas aseguradoras no tratan por considerarlo un tratamiento experimental.
En 1992 se rodó en EEUU El aceite de la vida,una película que cuenta la vida de Lorenzo y su familia, el descubrimiento de el aceite y cómo es la ALD (adrenoleucodistrofia).
“Soy padre. Y mi implicación en la enfermedad llamada
‘Adrenoleucodistrofia’ no viene, por tanto, del amor a la ciencia sino
del amor a mi hijo Lorenzo y de mi deseo de ayudarlo. Debía haber muerto
hace 13 años… y hoy tiene 24. Ciertamente, tiene días buenos y días
malos, está postrado y no puede comer más que a través de un tubo… pero
su mente sigue ahí. Le gusta que le leamos, que toquemos música para él y
sabe quién está a su alrededor. Estoy muy contento con los resultados del estudio. No porque hayan
elevado mi ego, ni debido a la publicidad que han generado. Estoy
contento porque el Aceite de Lorenzo ha salvado y salvará las vidas de muchos niños afectados
por Adrenoleucodistrofia. A pesar de que los médicos reconocieron muy
pronto que el Aceite de Lorenzo eliminaba el defecto bioquímico -es
decir, la acumulación de la cadena de ácidos grasos- la mayoría fue
escéptica sobre su valor terapéutico. Está claro que ese escepticismo se
debió al hecho de que ni mi esposa ni yo pertenecíamos a la comunidad
médica. En otras palabras, no creyeron posible que dos personas
completamente ajenas al mundo de la investigación, dos laicos de la
medicina, pudieran desarrollar una terapia para un desorden raro,
neurológico. Aunque la mayoría de los doctores sabe muy bien que su
comprensión del cuerpo humano es limitada algunos no lo asumen y se vuelven arrogantes.
Y creo que el problema no radica tanto en los médicos como en quienes
fuera de la medicina los sitúan en un pedestal. Afortunadamente, el
escepticismo de los médicos sobre el Aceite de Lorenzo ha disminuido ya y
disminuirá aún más a medida que se extiendan los resultados del
estudio.” El aceite de la vida
También os aconsejo ver la película. Es una historia que hace reflexionar ya que la mayoría de las enfermedades raras no tiene cura; sin embargo, unas personas que no tenían nada que ver con las ciencias lograron dar con la cura para la enfermedad de su hijo.
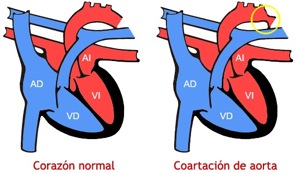
Hoy toca hablar sobre otro tipo de cardiopatía congénita, la coartación de la aorta. Esta patología se produce cuando hay un estrechamiento de la arteria aorta fuera del corazón. La causa normalmente es desconocida, pero se presenta con frecuencia en niñas con anormalidades cromosómicas como el Síndrome de Turner. Además, no se hereda genéticamente.
Si el estrechamiento es importante, la parte principal del corazón, que es el ventrículo izquierdo, tiene que trabajar más fuerte para impulsar la sangre a través de la arteria aorta, que está afectada.
Esta patología puede ser curada mediante una intervención quirúrgica, si el estrechamiento es severo y si se localiza en los primeros días de vida. Sin embargo, si se detecta en niños mayores de un mes, el neonato puede ser tratado mediante un cateterismo cardíaco a través de una angioplastia.
Por otro lado, si el estrechamiento es ligero, no precisa cirugía.
¿Cómo afecta a los niños?
Si el estrechamiento es severo, el niño se cansará fácilmente, tendrá la presión arterial elevada y no se sentirán los pulsos en ambas ingles.
Por el contrario, si es ligero no afectará al niño.
Es una enfermedad que se caracteriza en la deficiencia de la producción y síntesis de melanina que ocasiona poco o ningún pigmento en la piel, cabello y los ojos.
¿Cuáles son sus causas?
La síntesis de melanina se lleva a cabo por estructuras celulares especializadas llamadas melanocitos. Para que sintetice esta sustancia se llevan a cabo una serie de reacciones enzimáticas en cadena en las que intervienen otras enzimas de gran importancia como: fenilananina y tirosina, siendo la tetrahidrobiopterina un compuesto que limita la velocidad de la síntesis de melanina.
La tirosina, precursor de la melanina, se transporta al melanosona donde sufre una serie de pasos hasta transformarse en la melanina.
¿Qué síntomas presenta?
Una persona que presenta esta enfermedad tendrá:
Ausencia del color en el cabello y el iris y la retina del ojo
Pelo y piel más claro de lo normal
Y otras menos frecuentes como:
Ojos bizcos
Fotofobia o sensibilidad a la luz
Problemas de visión como puede ser la ceguera (que puede llegar a ser total)
¿Y su tratamiento?
El tratamiento es aliviar los síntomas y consiste en proteger la piel y los ojos, principalmente del sol:
Disminuir el riesgo de sufrir quemaduras debido al sol (la piel es mucho más sensible a la radiación), con la ayuda de protectores solares de alto nivel protector.