La mucolipidosis es una patología autosómica recesiva producida por el déficit de la enzima N-Acetilglucosamina - 1 - fosfotransferasa. Su déficit ocasiona que las hidrolasas ácidas ingresen a estos organelos siendo liberadas en exceso al medio extracelular conduciendo finalmente al daño tanto tisular como celular.
El déficit de esta enzima ocasiona 2 fenotipos: mucolipidosis tipo II (I - Cell) y la tipo III o polidistrofia pseudo-Hurler.

¿Que síntomas presenta?
La mucolipidosis tipo II es una patología severa cuyas manifestaciones se pueden observar desde el período neonatal.
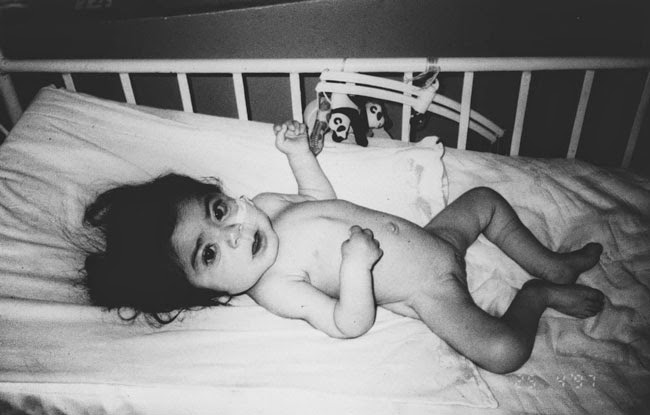
Su cuadro clínico se caracteriza por dismorfias faciales similares a la enfermedad de Hurler, junto a enfermedades esqueléticas, severo retraso psicomotor, deterioro psico-orgánico progresivo y muerte entre el primer y segundo año de vida.
Se observan también macrocefalia, cabello seco, piel gruesa, opacidad corneal, hipertrofia adenoides, hepatoesplenomegalia, manos en contractura de garra y hernias umbilical e inguinal, entre otros.
En la siguiente foto podemos observar una niña con dicha patología. Toda la información sobre este caso clínico lo podéis leer en el enlace que dejaré en el apartado "Para más información" de Scielo.
Para más información:
- FEDER:http://www.enfermedades-raras.org/index.php/las-enfermedades-raras/listado-de-patologias?id=1118
- SCIELO:http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0034-98872003000300011
David Rey Bretal
No hay comentarios:
Publicar un comentario